

Project leader:
Christian Rödelsperger
Department:
IV - Evolutionary Biology
Director:
Ralf. J. Sommer
Office:
Maria Gölz
Max-Planck-Ring 9
D-72076 Tübingen
Germany
Phone: +49 7071 601 440/441
Fax: +49 7071 601 498
How do processes like duplication, genomic rearrangements, and formation of novel genes shape genomes? Do these processes generate phenotypic differences that we care about? To extend our understanding of these two questions, we combine large-scale sequencing data with statistical analysis to find the genetic basis of various traits in the nematode P. pacificus. Nematodes such as P. pacificus and C. elegans are excellent systems to study these two questions, because they are easy to maintain in the lab, have small genomes, and have a broad range of well established experimental protocols including genome editing. Most importantly, nematodes exhibit numerous interesting morphological, developmental, and behavioral traits, of which the genetic basis and evolution is still poorly understood.
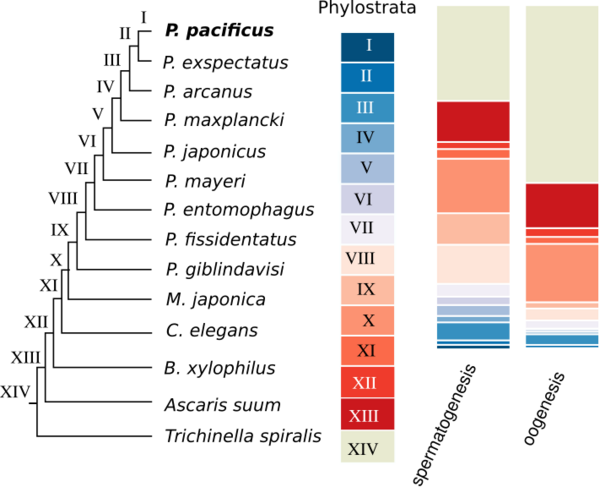
P. pacificus was among the first nematodes with a sequenced genome. Initial analysis showed that only around one third of genes do not have homologs in any other sequenced nematode genome. Such genes are called orphan genes. Since most existing functional annotations are inferred from homologs in classical model organisms, basically no functional data exists for orphan genes. One of our main goals is to understand the evolutionary dynamics and origin of orphan genes. We further perform large-scale gene expression profiling throughout development, different tissues, and varying environmental conditions to make identify potential functions of orphan genes.
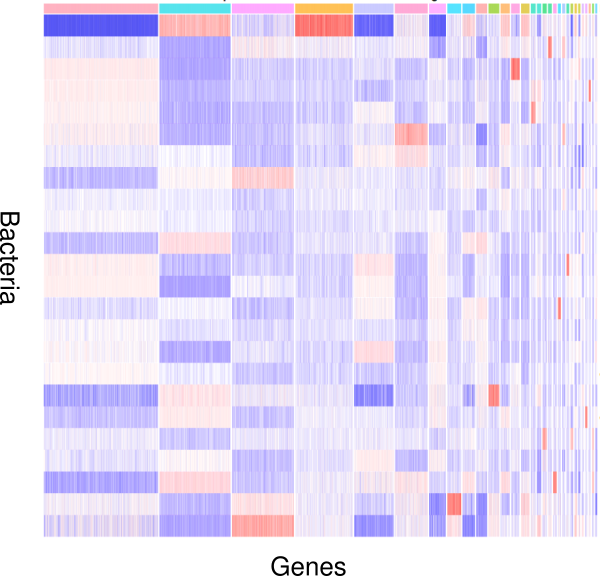
How do bacteria modulate nematode development and behavior? Which genes respond to changes in environmental microbiota? To study these questions, we apply an integrative approach involving genomic and transcriptomic data. Nematodes like P. pacificus and C. elegans are excellent models to study the interactions between bacteria and their hosts, because they are easy to grow using monoxenic bacterial cultures as a food source. In addition, worms as well as bacteria are genetically tractable, which can provide detailed mechanistic insights into the interaction between host and bacteria and their impact on development and behavior.
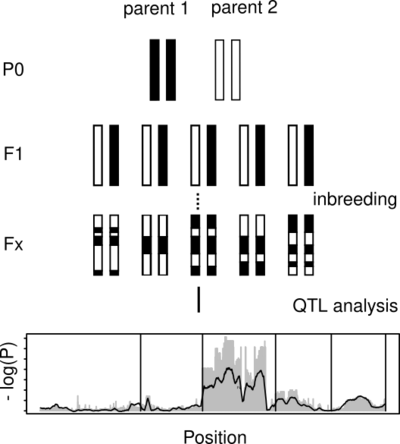
It is relatively easy to sequence a genome. However, understanding the function and biological significance of individual genes, still takes an enormous amount of experimental work. One major part of our work is to contribute to the identification of links between genotype and phenotype. In close collaboration with other scientists from our department, we work on identifying the genetic basis for various traits. This involves the analysis of whole-genome sequencing data from mutant strains, artificial crosses, and wild populations. The availability of efficient gene editing allows us to identify causal genes by knock out. Further comparative and population genomic characterization of the identified loci can give further insights into the evolution of the associated traits.
Alumni:
Athanasouli M & Rödelsperger C (2022): Analysis of repeat elements in the Pristionchus pacificus genome reveals an ancient invasion by horizontally transferred transposons.
BMC Genomics. 23:523. DOI: 10.1186/s12864-022-08731-1.
Athanasouli M, Witte H, Weiler C, Loschko T, Eberhardt G, Sommer RJ, & Rödelsperger C. (2020): Comparative genomics and community curation further improve gene annotations in the nematode Pristionchus pacificus. BMC Genomics. 21:708. DOI: 10.1186/s12864-020-07100-0.
Baskaran, P., Rödelsperger, C., Prabh, N., Serobyan, V., Markov, G. V., Hirsekorn, A. & Dieterich, C. (2015): Ancient gene duplications have shaped developmental stage-specific expression in Pristionchus pacificus. BMC Evol Biology. 15:185. DOI: 10.1186/s12862-015-0466-2.
Baskaran, P. & Rödelsperger, C. (2015): Microevolution of Duplications and Deletions and Their Impact on Gene Expression in the Nematode Pristionchus pacificus. PLoS One, 10:e0131136. DOI: 10.1371/journal.pone.0131136.
Baskaran, P., Jaleta, T.G., Streit, A. & Rödelsperger, C. (2017): Duplications and Positive Selection Drive the Evolution of Parasitism-Associated Gene Families in the Nematode Strongyloides papillosus. Genome Biol Evol. 9, 790-801. DOI: 10.1093/gbe/evx040.
Prabh,N. & Rödelsperger, C. (2016): Are orphan genes protein-coding, prediction artifacts, or non-coding RNAs? BMC Bioinformatics. 17, 226, DOI: 10.1186/s12859-016-1102-x.
Prabh, N. Röseler, W., Witte, H., Eberhardt, G., Sommer, R.J. & Rödelsperger C. (2018): Deep taxon sampling reveals the evolutionary dynamics of novel gene families in Pristionchus nematodes. Genome Research. 28, 1664-1674. DOI: 10.1101/gr.234971.118.
Prabh,N. & Rödelsperger, C. (2019): De Novo, Divergence, and Mixed Origin Contribute to the Emergence of Orphan Genes in Pristionchus Nematodes. G3. 9, 2277-2286. DOI: 10.1534/g3.119.400326.
Prabh N & Rödelsperger C. (2022): Multiple Pristionchus pacificus genomes reveal distinct evolutionary dynamics between de novo candidates and duplicated genes. Genome Research. 32, 1315-1327. DOI: 10.1101/gr.276431.121.
Rödelsperger C, Röseler W, Prabh N, Yoshida K, Weiler C, Herrmann M & Sommer RJ. (2018): Phylotranscriptomics of Pristionchus Nematodes Reveals Parallel Gene Loss in Six Hermaphroditic Lineages. Curr Biol. 28:3123-3127.e5. DOI: 10.1016/j.cub.2018.07.041.
Rödelsperger C , Prabh N. & Sommer R.J (2019): New Gene Origin and Deep Taxon Phylogenomics: Opportunities and Challenges. Trends Genet. 35:914-922. DOI: 10.1016/j.tig.2019.08.007.
Rödelsperger C. , Annabel Ebbing A , Sharma D.R. , Okumura M, Sommer R.J. & Korswagen H.C (2021): Spatial Transcriptomics of Nematodes Identifies Sperm Cells as a Source of Genomic Novelty and Rapid Evolution. Molecular Biology Evolution. 38:229-243. DOI: 10.1093/molbev/msaa207.
